
January/February 2018
Medication Monitor: Biosimilars Can Reduce Drug Costs
By Mark D. Coggins, PharmD, BCGP, FASCP
Today's Geriatric Medicine
Vol. 11 No. 1 P. 5
Biological products or biologics have revolutionized the treatment of many serious and chronic illnesses and are the fastest growing class of therapeutic products in the United States. Biologics have been life changing for many patients suffering from a wide range of conditions including rheumatoid arthritis, certain cancers, diabetes, anemia, low white blood cell counts, macular degeneration, multiple sclerosis, inflammatory bowel disease, and skin conditions such as psoriasis.
The High Cost of Biologics1
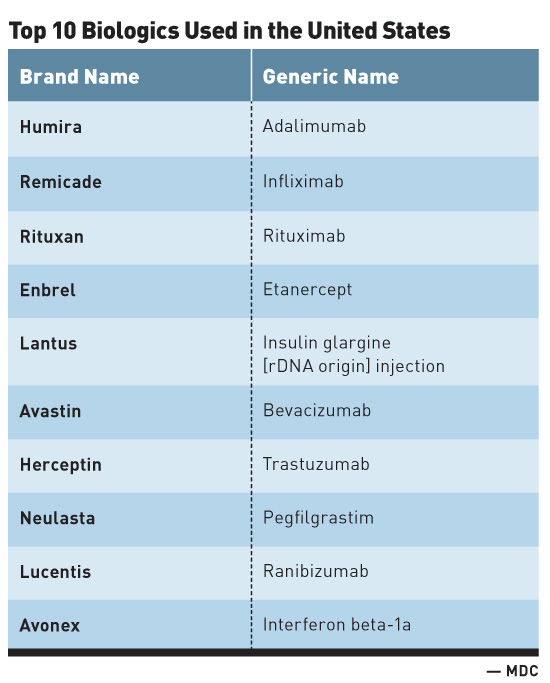
The tremendous advancements seen with biologics are not without significant financial cost. For instance, while biologics account for less than 1% of all dispensed prescription drugs in the United States, they account for one-fourth of all prescription spending. The anti-inflammatory biologic Humira (adalimumab) manufactured by AbbVie is the most commonly used biologic in the United States. With global sales of $14 billion in 2015, Humira is also the best-selling drug worldwide regardless of drug class. Additionally, the top 10 biologics used in the United States (see table) are all designated as blockbuster drugs, denoting that each has generated much more than $1 billion in annual sales.
Biologics Price Competition and Innovation Act2
Congress approved the Biologics Price Competition and Innovation (BPCI) Act of 2009 to help increase access to high-cost lifesaving biologics and increase the development of more treatment options, and in an effort to potentially lower health care costs through competition. This legislation created a licensure pathway for biological products that are demonstrated to be biosimilar to or interchangeable with an FDA-approved biological product.
Introduction of Biosimilars in the United States
The BPCI Act laid the groundwork for the development and subsequent approval of biosimilars, also sometimes referred to as biogenerics.2 In 2015, the US patent for Rituximab that was first issued in 1998 expired. As a result, biologics began facing competition in the United States when IDEC Pharmaceuticals developed a biosimilar product for Rituximab. Since 2015, additional biologics have lost patent protection, including Humira in 2016 and Remicade, which will lose its protections in 2018. A larger number of top biologics will lose patent exclusivity by 2020, making it likely that there will be an accelerated availability of biosimilars.1
Confusion Surrounding Biosimilars2
The emergence of biosimilars has resulted in confusion among many health care providers and payers as to what biosimilar products are and how their use may impact the provision of care and pharmaceutical costs moving forward. As a result, the FDA and biosimilar manufacturers have spent considerable time and effort in working to increase the education surrounding these products.
Biosimilars Defined
A biosimilar is a biological product that is highly similar to and has no clinically meaningful differences from an existing FDA-approved reference product.
When a biosimilar is being submitted for approval by the FDA, the expectation is that the biosimilar will work in a way that is highly similar to an already approved biologic medicine known as the originator biologic or reference product. For instance, using Product A as an example of an approved biologic reference product, when Product A seeks approval, the FDA evaluates and approves its use using, among other things, a full complement of safety and effectiveness data. After a period of time, Product A's (originator biologic/reference product) patent expires and a manufacturer seeking approval for a new biosimilar, Product B, brings it to the FDA for evaluation and subsequent approval.
Demonstrating "Highly Similar"
A manufacturer developing a proposed biosimilar demonstrates that its product is highly similar to the reference product by extensively analyzing (ie, characterizing) the structure and function of both the reference product (eg, approved biologic Product A) and the proposed biosimilar (eg, proposed biosimilar Product B). State-of-the-art technology is used to compare characteristics of the products, such as purity, chemical identity, and bioactivity. Minor differences between the reference product and the proposed biosimilar product in clinically inactive components are acceptable. For example, these could include minor differences in the stabilizer or buffer compared with what is used in the reference product. The FDA carefully evaluates any differences between the proposed biosimilar product and the reference product to ensure the biosimilar meets the FDA's high approval standards.
Highly Similar Does Not Mean Exact Replica
Biological products are generally large complex molecules vs most of the medications that have been brought to the market over the years, which are called small molecule drugs, such as aspirin. Whereas small molecule drugs are relatively easily made using chemicals in a lab, biologics—their biosimilar counterparts—are often produced through biotechnology (recombinant DNA technology) in a living system, such as a microorganism or plant or animal cell, and are often more difficult to characterize than small molecule drugs. As mentioned previously, slight differences (ie, acceptable within-product variations) are expected during the manufacturing process for biological products, regardless of whether the product is a biosimilar or a reference. Given the complexity of this process, it's unlikely for biosimilars to be exact replicas of the reference product. An important point is that even when comparing lots of a reference biologic product to itself, slight differences between manufactured lots of the same biological product (ie, acceptable within-product variations) are normal and expected within the manufacturing process. In other words, even when comparing one batch of a reference product to another batch of the same reference product, it is very unlikely that the products will be exactly the same.
No Clinically Meaningful Differences
In addition to a proposed biosimilar being required to be highly similar to the original reference product, a manufacturer must demonstrate that its proposed biosimilar product has no clinically meaningful differences from the reference product in terms of safety, purity, and potency (safety and effectiveness).
This is generally demonstrated through human pharmacokinetic (exposure) and pharmacodynamic (response) studies, an assessment of clinical immunogenicity and, if needed, additional clinical studies.
Interchangeable Biosimilars
Additional requirements outlined by the BPCI Act are required for a particular biosimilar product to be deemed interchangeable with its reference product. An interchangeable biologic may be substituted (where allowed by state law) for the reference biologic without the involvement of the prescriber.
The FDA has high standards for approval, which should assure health care providers that they can be confident in the safety and effectiveness of an interchangeable product, just as they would be for an FDA-approved reference product.
FDA-approved interchangeable biosimilars have been shown to produce the same clinical result as the reference product in any given patient. Also, for biosimilars administered to a patient more than once, the risk in terms of safety and reduced efficacy of switching back and forth between an interchangeable product and a reference product will have been evaluated.
For example, a patient may self-administer a biological product by injection to treat rheumatoid arthritis. To receive the biosimilar instead of the reference product, the patient may need a prescription from a health care prescriber written specifically for that biosimilar.
However, once a product is approved by the FDA as interchangeable, the patient may be able to take a prescription for the reference product to a pharmacy and, depending on state pharmacy laws, a pharmacist could substitute the interchangeable biosimilar for the reference product without consulting the prescriber. The FDA undertakes a rigorous and thorough evaluation to ensure that all products, including biosimilar and interchangeable products, meet the FDA's high standards for approval.
Biosimilars Are Not Generic Drugs
Confusion often exists regarding biosimilars vs generic drugs. And while both share some similar characteristics, they are not the same. Similarities include the following:
• Biosimilars and generic drugs are versions of brand name drugs and may offer more affordable treatment options to patients.
• Biosimilars and generics are approved through different abbreviated pathways that avoid duplicating costly clinical trials.
However, important differences exist between biosimilars and generic drugs. Differences include the following:
• The active ingredients of generic drugs are exactly the same as those of brand name drugs.
• The manufacturer of a generic drug must demonstrate that the generic is bioequivalent (the generic and brand are for all intents and purposes the same) to the brand name drug.
Also, unlike generic drugs, which are chemically synthesized, biosimilars are grown in complex living systems. For both reference product and biosimilars, the FDA assesses the manufacturing process and the manufacturer's strategy to control within-product variations. These control strategies are put in place to help ensure manufacturers produce biologics, whether a reference product or biosimilar, with consistent clinical performance.
The "Purple Book"
The "Purple Book" lists biological products, including any biosimilar and interchangeable biological products, licensed by the FDA under the Public Health Service Act (the PHS Act).
The Purple Book includes the date a biological product was licensed under 351(a) of the PHS Act and whether the FDA evaluated the biological product for reference product exclusivity under section 351(k)(7) of the PHS Act.2
Potential Cost Savings
As the number of approved biosimilars increases, it will be increasingly important for health care providers to be able to understand and explain the differences of biosimilars to patients. Access to these biological products is challenging due to their high cost. The exact savings anticipated by using the biosimilar version of a reference biologic is difficult to predict. However, we do know that biosimilars cost about $75 million to $250 million to reach the approval stage vs around $2 million to $3 million for a generic small molecule medicine. Much like generics, biosimilars can help cut drug costs though the savings are smaller because of their complexity and regulatory challenges related to obtaining FDA approvals. Regardless, even savings that may be 20% less than a reference product can have a significant impact on the overall cost of these medications and help expand access to more patients.
— Mark D. Coggins, PharmD, BCGP, FASCP, is vice president of pharmacy services and medication management for Diversicare, which operates skilled nursing centers in 10 states. He was nationally recognized by the Commission for Certification in Geriatric Pharmacy with the 2010 Excellence in Geriatric Pharmacy Practice Award.
References
1. Stone K. Top 10 biologic drugs in the United States. The Balance website. https://www.thebalance.com/top-biologic-drugs-2663233. Updated October 12, 2017. Accessed December 4, 2017.
2. Biosimilars. FDA website. https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsare
developedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/default.htm. Updated November 14, 2017. Accessed December 4, 2017. |